研究 (Research)
最終更新日:
データ駆動型界面反応過程シミュレーションの開発と応用 (Development and application of data-driven simulation techniques for interface chemical processes)
教授 森川 良忠(工学研究科 物理学系専攻) MORIKAWA Yoshitada(Graduate School of Engineering)
研究の概要
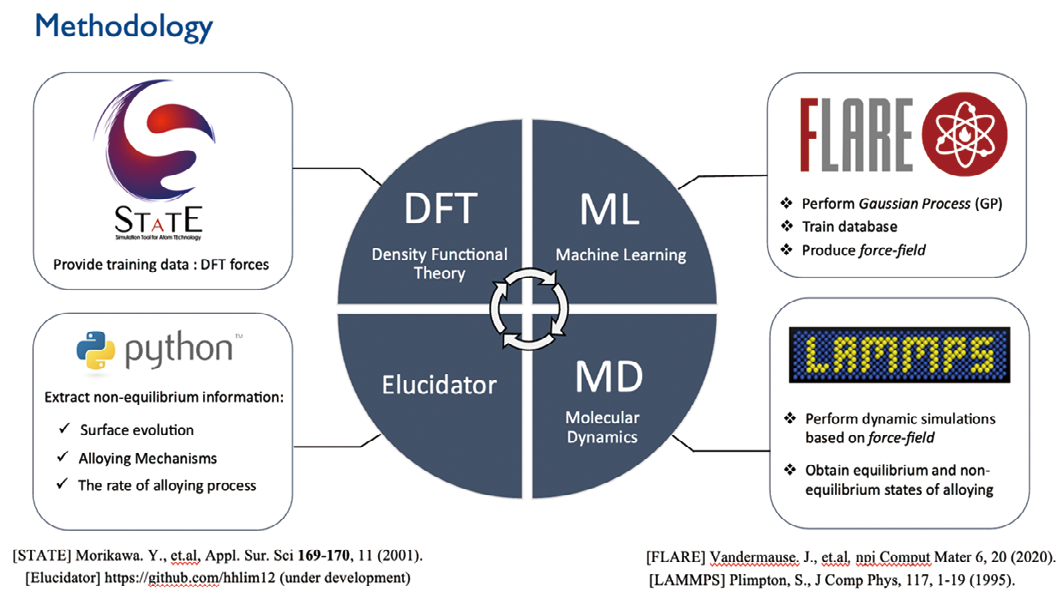
界面での構造や電子状態、化学反応過程を原子レベルで解明することは基礎科学的のみならず、応用上も極めて重要である。密度汎関数理論 (DFT)は原子・分子レベルでの構造や電子状態を極めて精度良く計算することが可能であるが、その計算は重く、取り扱える系の空間サイズは数 nm、時間は数十 ps 程度に限られる。一方、経験的な原子間ポテンシャルを用いると、サブ µm の空間サイズ、µs の時間スケールの現象の解明が可能となる。DFT を機械学習法によりフィットした機械学習原子間ポテンシャルを構築することにより、DFT に近い精度でより大きなスケールのシミュレーションが可能になる。本研究ではこの手法を用いてさまざまな界面反応の解明を行い、より望ましい界面反応のための設計指針を与えることを目指す。
研究の背景と結果
気体 / 固体、液体 / 固体、あるいは固体 / 固体界面での構造や電子状態、化学反応過程を原子レベルで解明することは、基礎科学的のみならず、不均一触媒や半導体デバイス、結晶成長、エッチング、燃料電池、二次電池や摩耗、潤滑などさまざまな応用分野で重要である。しかしながら、それらを実験的に解明することは困難な場合が多い。そのため、計算精度が高い DFT 計算による原子レベルでの解明が極めて重要な役割を果たす。しかしながら、その計算量は膨大であるため、計算できる系の空間スケールは数 nm 程度、時間スケールは数十 ps 程度に限られていた。我々は DFT の計算結果を機械学習法により原子間力ポテンシャルにフィットすることにより、高精度な計算を非常に高速に行うことを可能にし、それを用いてメタノール合成触媒反応で重要なCu-Zn 表面合金形成過程の解明を行った。機械学習ポテンシャルにより空間スケールがサブ µm の空間、時間スケールが ms の分子動力学シミュレーションを行うことにより、合金形成過程の素過程を明らかにした。
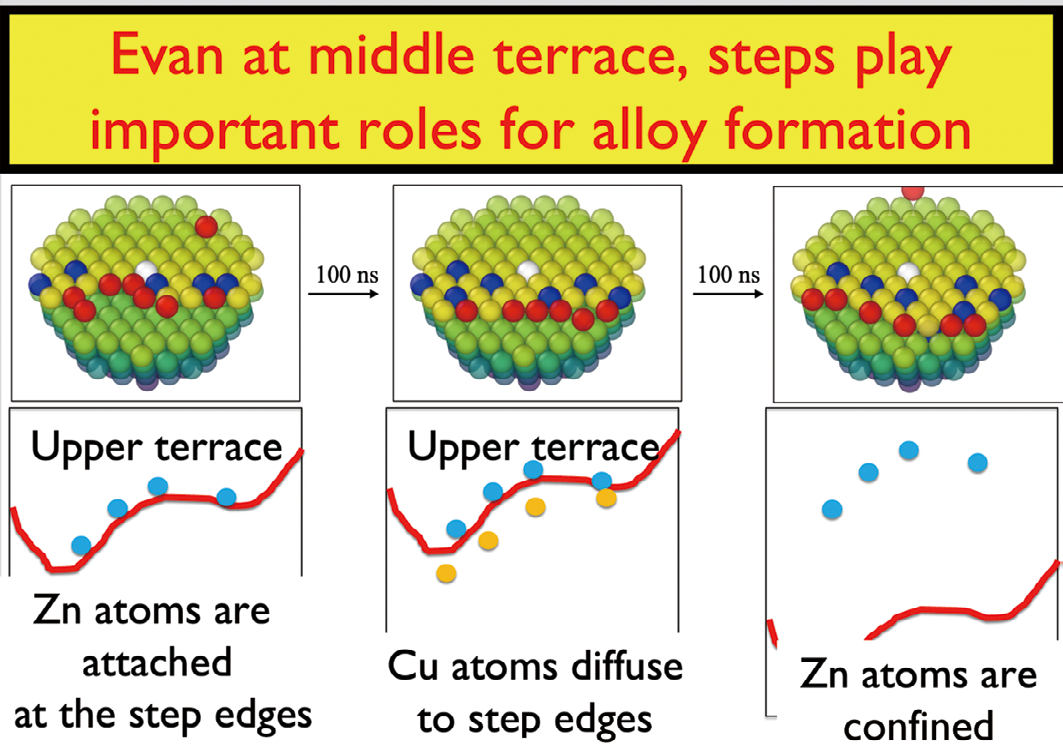
この結果、固体表面上の原子ステップ付近でステップ端上側の原子と下側の原子の間で非常に頻繁に原子の交換が行われて合金化が進行することを明らかにした。これは、STM を用いて観測された結果と定性的には一致するが、実験的に提案されていた原子交換による合金形成とはかなり異なった原子過程が頻繁に起こっていることが明らかとなった。このように機械学習ポテンシャルによるシミュレーションが界面での原子レベルでの素過程を解明する上で極めて有効であることを示した。
この有効な手法を、加工ツールの材料として重要で、さらには次世代の半導体として期待されるダイヤモンドの表面酸化エッチング過程やそのほか重要な触媒や燃料電池電極反応過程などさまざまな系に適用して行く予定である。
研究の意義と将来展望
精度の高い DFT は極めて大きな役割を果たしてきているが、計算は大変重いため、最新鋭のスパコンを用いたとしても、限られた仮定のモデルを用いる必要があった。最近の機械学習法の進展は、DFT によるさまざまな構造に対する計算結果から、精度の高い原子間ポテンシャルを導き出すことを可能にする。より現実デバイス環境下での界面の反応シミュレーションが可能になり、将来的には反応過程の予測やより望ましい物質の設計が可能になると期待できる。
担当研究者
教授 森川 良忠(工学研究科 物理学系専攻)
キーワード
密度汎関数理論/機械学習/表面科学/不均一触媒/電気化学
応用分野
エネルギー/環境/新材料